[Go to Japanese page]
Viewer of model and template 3D structures
We employ the common interface program "3D model viewer"(model3D.cgi) for displaying 3D structure of complex 3D structures and models provided by HOMCOS server.
When you click the icon  in search results of the HOMCOS serice, the 3D model viewer is shown.
in search results of the HOMCOS serice, the 3D model viewer is shown.
A folloing figure is the 3D model viewer for the model of CDKL1_HUMAN and CCNB1_HUMAN using 5hq0 as a template, by the service "Modeling Complex 3D Strucutreof Hetero Protein Multimer".
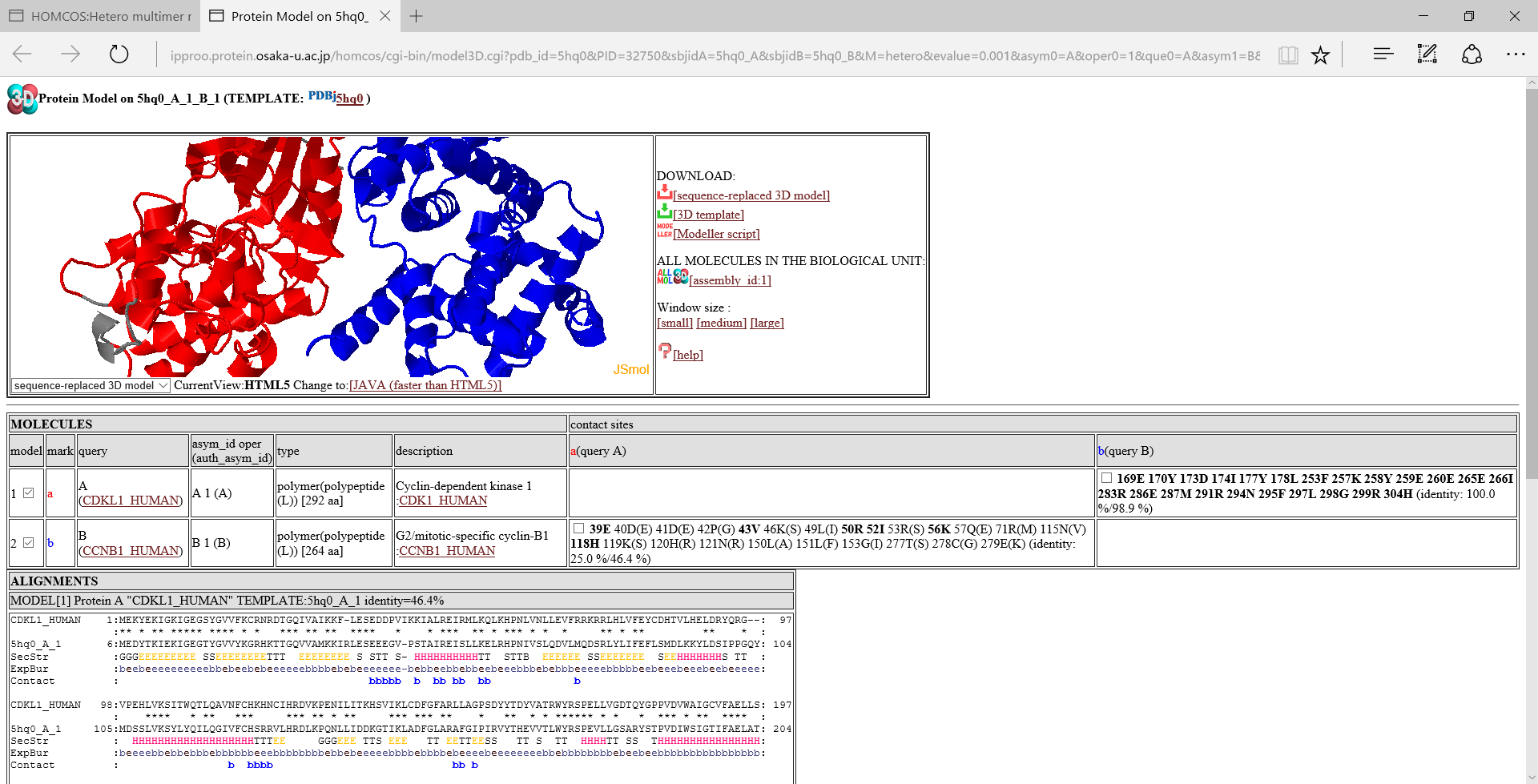
Icons in the left

 [sequence-replaced 3D model]:download the sequence-replaced 3D model.
This model is created simply by replacing the residue names and numbers of the template, with those of the query protein according to the BLAST alignment. The model can be generated with much less computation time than a full atomic modeling because it just replaces strings of the PDB file. However it is sufficiently useful for observing its molecular geometry at residue-level resolution. Of course, it cannot be used for docking and molecular dynamics simulation, because its substituted side chains are not correctly modeled and all of the inserted residues are missed. For users who desire more detailed models, the HOMCOS server provides input files for MODELLER program.
[sequence-replaced 3D model]:download the sequence-replaced 3D model.
This model is created simply by replacing the residue names and numbers of the template, with those of the query protein according to the BLAST alignment. The model can be generated with much less computation time than a full atomic modeling because it just replaces strings of the PDB file. However it is sufficiently useful for observing its molecular geometry at residue-level resolution. Of course, it cannot be used for docking and molecular dynamics simulation, because its substituted side chains are not correctly modeled and all of the inserted residues are missed. For users who desire more detailed models, the HOMCOS server provides input files for MODELLER program.
 [3D template]:download the template PDB file.
[3D template]:download the template PDB file.
 [Modeller script]:download files to execute the Modeller program. Three files will be generated: an alignment file, an input python file, a template PDB file. We will explain elsewhere about how to execulete Modeller.
[Modeller script]:download files to execute the Modeller program. Three files will be generated: an alignment file, an input python file, a template PDB file. We will explain elsewhere about how to execulete Modeller.
- Window size [small] [medium] [large] :Change the windows size of the molecular viewer.
- ALL MOLECULES IN THE BIOLOGICAL UNIT

 [assembly_id:1]:If you click this icon, then the page is reloaded to include all of the molecules in the biological unit with the specific assembly_id. In the model linked direcrly from the search page, only two molecules are often included. However, other molecules are sometimes included in the biological unit, and they may be useful for disussing the function of the modeled structure. A following image is an example of "ALL MOLECULES".
[assembly_id:1]:If you click this icon, then the page is reloaded to include all of the molecules in the biological unit with the specific assembly_id. In the model linked direcrly from the search page, only two molecules are often included. However, other molecules are sometimes included in the biological unit, and they may be useful for disussing the function of the modeled structure. A following image is an example of "ALL MOLECULES".
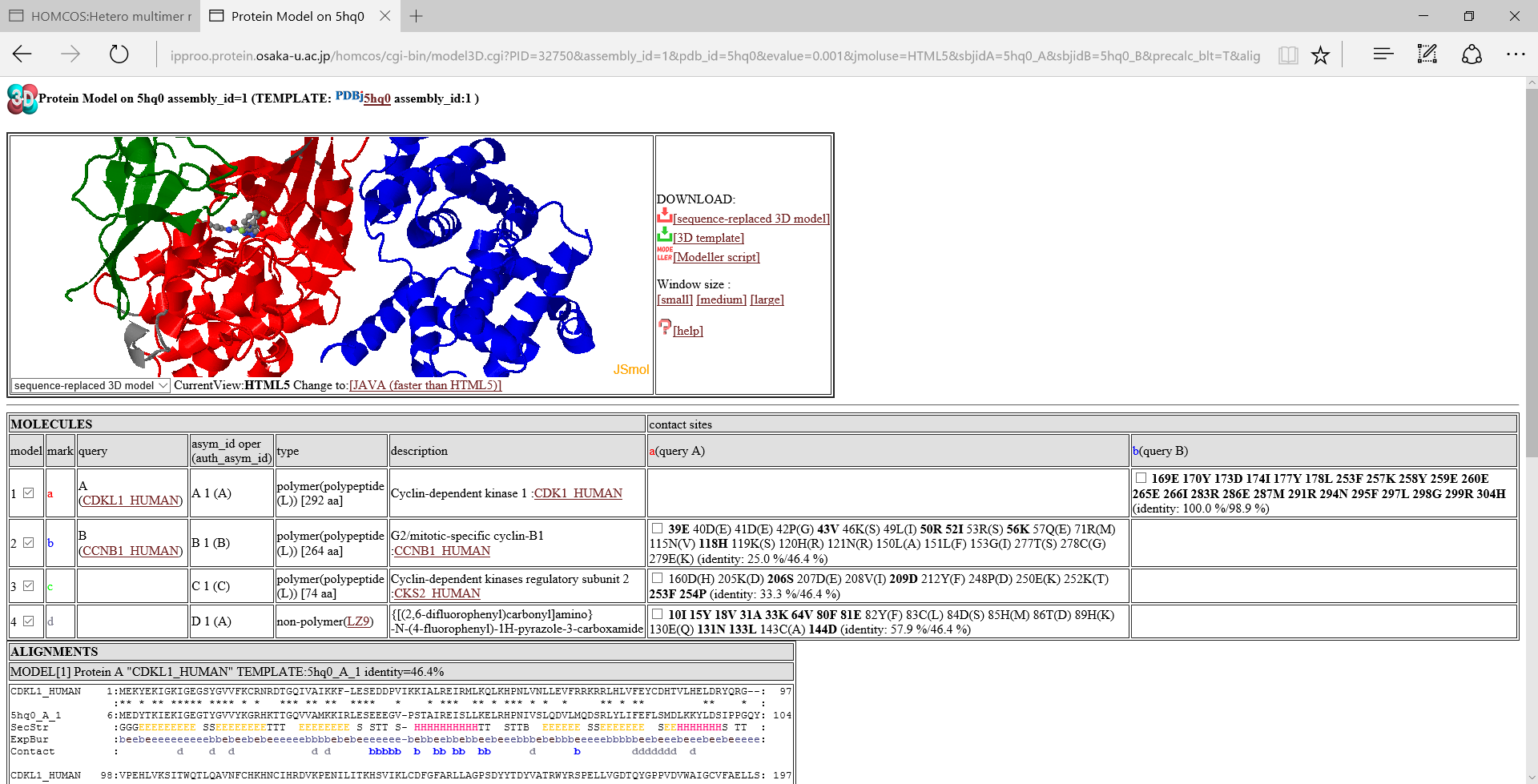
Table of molecules

- model:model number. A check box can change the displaying status of the molecule.
- mark:An alphabet representing the molecule. It is used to showing contacting sites in the alignment.
- query:type of query. If two proteins are used as the query, a symbol "A" or "B" will be shown. If possible, UniProt ID is shown.
- asym_id oper (auth_asym_id):this column shows asym_id, oper_expression, auth_asym_id of the molecule.
- type:type of the molecule. If the molecule is a protein, type is "polymer(polypeptide(L))" with number of amino acids. If the molecule is a compound, it shows three-letter code.
- description:a description (name) of the molecule. If possible, UniProt ID is shown.
- contact sites::Contact sites of query proteins are shown by their residue numbers and residue names. For example, the column "a(query A)" shows contact sites of the query protein A for the molecules, using the molecule a as the template. The bold sites, such as 39E are sites with common amino acids between the template and the query. Two identity values, such as "(identity: 25.0 %/46.4 %)" are squence identity. The first value ("25.0 %") is the identity only for the contact sites, whereas the second one ("46.4%") is the identity for the entire protein. A check box can change the displaying status of the contact sites.
Alignment
 Alignments of the query protein and the template protein are shown. These alignments are basicaly generated by the BLAST program. The line "SecStr" is for secondary structure of the template, the line "ExpBur" is for exposed("e") or buried("b") states of the template. The line "Contact" shows contact sites of the molecule. For example, the alphabet b shows a contact site with the molecule b, that is indicated in the "mark" column in the table of molecules.
Alignments of the query protein and the template protein are shown. These alignments are basicaly generated by the BLAST program. The line "SecStr" is for secondary structure of the template, the line "ExpBur" is for exposed("e") or buried("b") states of the template. The line "Contact" shows contact sites of the molecule. For example, the alphabet b shows a contact site with the molecule b, that is indicated in the "mark" column in the table of molecules.
Comments and Questions to:


 3D model view
3D model view